maj 16, 2023
Philips provides update on completed set of test results for CPAP/BiPAP sleep therapy devices
Amsterdam, the Netherlands – Royal Philips (NYSE: PHG; AEX: PHIA), today provides an update on the comprehensive test and research program of its subsidiary Philips Respironics to assess potential health risks related to the polyester-based polyurethane (PE-PUR) sound abatement foam in specific sleep therapy and ventilator devices under the voluntary June 2021 recall notification/field safety notice.*
The risk assessments have now been completed for the CPAP/BiPAP sleep therapy devices under the recall notification/field safety notice* i.e., the first-generation DreamStation, System One and DreamStation Go devices, representing approximately 95% of the registered devices globally. The assessments build on the previous updates in December 2021, June 2022, and December 2022. Additionally, tests and analyses have been completed for first-generation DreamStation devices that have been exposed to ozone cleaning.
Test methods
The test and research program has been conducted together with five independent, certified testing laboratories, and the results have been reviewed and assessed by third-party qualified experts and Philips Respironics, as well as an external medical panel.
The applied test methods – comprising test planning, test execution, and interpretation of the results for the completed risk assessments – are in accordance with the applicable ISO 18562 [1,2] and ISO 10993 [3] industry standards. The design of the applied test methods was further scientifically underpinned based on a thorough consideration and mitigation of inherent testing limitations. For example, testing was performed on multiple Used devices with differing amounts of patient usage and observed visual foam degradation, and on Lab-Aged foam that had been intentionally degraded to different degrees. Very conservative assumptions were included in the risk assessments. Further examples are provided at the bottom of this press release.
“Our first priority is the health and well-being of patients,” said Roy Jakobs, CEO of Royal Philips. “We have therefore focused on the comprehensive test and research program to gain more clarity about the safety of the affected devices, and on providing replacement devices to patients. The third-party risk assessments for the sleep therapy devices presented today are positive and reassuring, and we are making good progress with the remediation of the affected devices. The relevant competent authorities globally, including the FDA, are still reviewing the test results and assessments. We share the same objective to ensure patient safety and quality in the delivery of healthcare, and we remain committed to working closely with these agencies. The completion of testing and remediation of the affected devices remain our highest priorities.”
Test results and analyses for sleep therapy devices not exposed to ozone cleaning
The completed set of test results and analyses for the first-generation DreamStation, System One and DreamStation Go sleep therapy devices indicate that potential patient exposure to foam particulate matter (PM) and volatile organic compounds (VOCs) from the PE-PUR foam within these devices is unlikely to result in an appreciable harm to health in patients. The conducted tests and conclusions have been summarized in the table below.
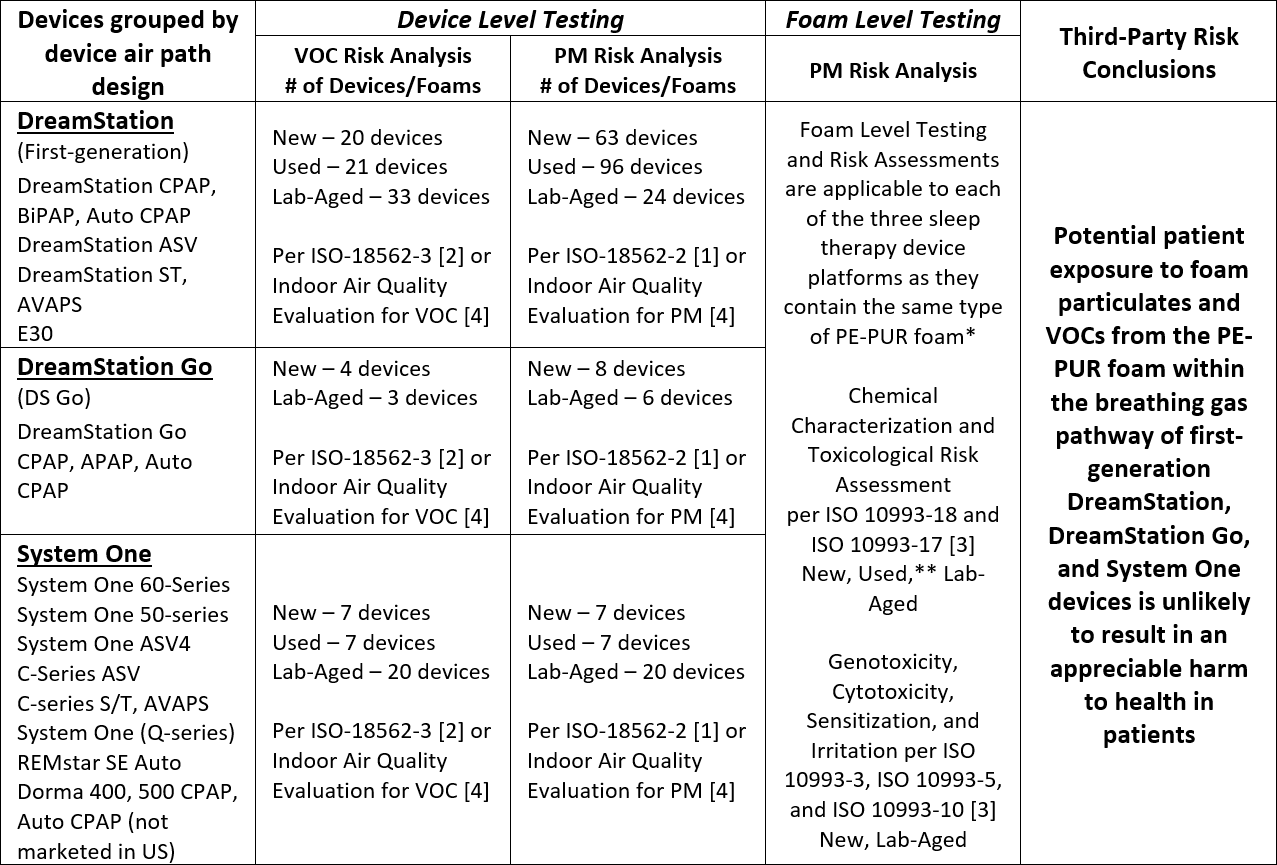
VOC: Volatile Organic Compound
PM: Particulate Matter
* The total amount of foam in the devices varies from approximately 1 g to 10 g, depending on the device airpath design and configuration. Devices within each platform share the same airpath design and configuration, including the amount of foam present.
** The foam from 7 different Used first-generation DreamStation devices was chemically characterized per ISO 10993-18 and -17 [3] and included foam representative of a range of visual degradation states.
Expanded testing and toxicological risk assessments on multiple devices with New, Used, and Lab-Aged foam have shown no appreciable harm to health for the VOCs detected based on ISO 18562-3 [2] testing, and a third-party risk assessment concluded that exposure to VOCs for these devices is unlikely to result in an appreciable harm to health in patients.
New, Lab-Aged and Used devices were tested and all were compliant with ISO 18562-2 [1] allowable limits for PM emissions. Testing was performed on Used devices (i.e., devices previously used by patients) with varying degrees of degradation (i.e., from no visible degradation to severe visible degradation), New devices, and devices with Lab-Aged foam that intentionally had been exposed to significantly elevated temperatures (≥80°C) and humidity (≥75% RH) to accelerate degradation.
Further, tested PM emissions of Used devices with visible degradation were not statistically different than PM emissions of Used devices without degradation, suggesting that degradation did not contribute to appreciable elevated levels of respirable particles in the devices tested.
Even with the very conservative and theoretical assumption that all of the foam could degrade and that a patient is then exposed to all of the degraded PE-PUR foam within the devices, the third-party risk assessment concluded that exposure to particulates from degraded foam in these devices, including potential respirable and non-respirable particulates, is unlikely to result in an appreciable harm to health in patients.
Based on the visual inspection of the foam in returned first-generation DreamStation devices, the prevalence of visible foam degradation was found to be low. Visual inspection can only identify visible foam degradation and cannot measure volatile organic compounds generation or quantify particulate loss, therefore additional testing and analyses were performed as described above and in the complete update.
The impact of ozone cleaning on foam degradation in first-generation DreamStation devices
Philips Respironics has completed testing on first-generation DreamStation devices that have been exposed to ozone cleaning:
ISO 18562-3 VOC testing showed that after 200 ozone cleaning cycles - each cycle simulating one night of use and then ozone cleaning - diethylene glycol (DEG) became detectable as a VOC. The testing was conducted up to 500 ozone cleaning cycles, and a VOC toxicological risk of this ozone-induced degradation determined that exposure to VOC emissions from the assessed first-generation DreamStation devices exposed to ozone cleaning suggests no appreciable risk to health for patients.
Regarding risks associated with respirable and non-respirable particulates, testing to date has been performed on devices with known ozone exposure. For example, two Used first-generation DreamStation devices with user-reported ozone exposure were included in extractables and leachables testing, which formed the foundation for a toxicological risk assessment of the sleep therapy device PE-PUR foam particulate. That third-party collective analysis concluded that exposure to particulate matter from degraded foam in the first-generation DreamStation devices is unlikely to result in an appreciable harm to health in patients.
As previously published, data for first-generation DreamStation indicates that devices with user-reported ozone cleaning are 14 times more likely to have significant visible foam degradation/volume reduction compared to devices with no user-reported ozone exposure. This observation is consistent with laboratory testing, where first-generation DreamStation devices exposed to increasing cycles of ozone cleaning had increasingly more severe visual degradation. However, as concluded above, this is unlikely to result in an appreciable harm to health in patients.
Summary of ongoing tests
Philips Respironics is in the process of completing various remaining tests and analyses. The risk assessments for System One and DreamStation Go devices (that contain the same foam as the first-generation DreamStation devices) treated with ozone cleaning are being completed. For the Trilogy 100/200 and OmniLab Advanced Plus ventilator devices, VOC and PM testing continues, as well as chemical evaluation and toxicological risk assessment. These devices contain a different type of PE-PUR foam than the first-generation DreamStation devices [5]. Philips Respironics expects to provide an update on this in Q3 2023.
Guidance for healthcare providers and patients
Patients currently using an affected sleep therapy device that has not been remediated and not registered yet, are requested to register their devices to facilitate the remediation.
Philips Respironics continues to advise patients using affected sleep therapy devices that have not been remediated yet to contact their physician or care provider to decide on a suitable treatment for their condition, which may include stopping use of their device, continuing to use their affected device, using another similar device that is not part of the recall, or using alternative treatments for sleep apnea. Moreover, patients are advised to follow Philips Respironics’ instructions and recommended cleaning and replacement guidelines for their sleep therapy device and accessories. Ozone and UV light cleaning products are not currently approved cleaning methods for sleep therapy devices or masks and should not be used.
Philips Respironics also continues to advise users of ventilator devices to contact their healthcare providers before making any changes to their therapy.
Scientific underpinning of the test methods
The design of the applied test methods was scientifically underpinned based on a thorough consideration and mitigation of testing limitations that are inherent to any test standard and/or scientific research. To illustrate this, examples of such considerations and mitigations have been listed below.
The scientific underpinning of the applied test methods included a thorough consideration and mitigation of testing limitations, for example:
Notes
* Voluntary recall notification in the US/field safety notice for the rest of the world.
[1] ISO 18562-2: Biocompatibility evaluation of breathing gas pathways in healthcare applications – Part 2: Tests for emissions of particulate matter.
[2] ISO 18562-3: Biocompatibility evaluation of breathing gas pathways in healthcare applications – Part 3: Tests for emissions of volatile organic compounds.
[3] ISO 10993: Biological evaluation of medical devices; Part 1: Evaluation and testing within a risk management process; Part 3: Tests for genotoxicity, carcinogenicity and reproductive toxicity; Part 5: Tests for in vitro cytotoxicity; Part 10: Tests for irritation and skin sensitization; Part 17: Establishment of allowable limits for leachable substances; Part 18: Chemical characterization of medical device materials within a risk management process.
[4] The standard that was used for tests prior to ISO 18562.
[5] First-generation DreamStation, System One and DreamStation Go devices contain Type A PE-PUR foam, while Trilogy 100/200 devices contain Type B PE-PUR foam, and OmniLab Advanced Plus devices contain Type A and Type B PE-PUR foams. The known differences between the Type A and Type B foams are that Type B foam can be used with an acrylic pressure sensitive adhesive, has a lower density, has a different thickness, and also contains an additive to reduce potential flammability.
About Royal Philips
Royal Philips (NYSE: PHG, AEX: PHIA) is a leading health technology company focused on improving people's health and well-being through meaningful innovation. Philips’ patient- and people-centric innovation leverages advanced technology and deep clinical and consumer insights to deliver personal health solutions for consumers and professional health solutions for healthcare providers and their patients in the hospital and the home. Headquartered in the Netherlands, the company is a leader in diagnostic imaging, ultrasound, image-guided therapy, monitoring and enterprise informatics, as well as in personal health. Philips generated 2022 sales of EUR 17.8 billion and employs approximately 74,000 employees with sales and services in more than 100 countries. News about Philips can be found at www.philips.com/newscenter.
Forward-looking statements
This statement contains certain forward-looking statements with respect to the financial condition, results of operations and business of Philips and certain of the plans and objectives of Philips with respect to these items. Examples of forward-looking statements include statements made about the strategy, estimates of sales growth, future EBITA, future developments in Philips’ organic business and the completion of acquisitions and divestments. By their nature, these statements involve risk and uncertainty because they relate to future events and circumstances and there are many factors that could cause actual results and developments to differ materially from those expressed or implied by these statements.
Topics
Contacts

Lisa Pernbrink
Philips Nordics Brand & Communication E-post: lisa.pernbrink@philips.com
You are about to visit a Philips global content page
ContinueMedia assets
")
")
Share on social media
Related news
-
![Philips i Almedalen: Panelsamtal om AI i vården]()
juni 02, 2025
-
![Philips på Vitalis 2025]()
maj 12, 2025
-
![Meet Philips at NCR 2025, May 21-23]()
maj 05, 2025